
天然产物靶向AR-铁死亡轴:抵抗前列腺癌耐药新策略


前列腺癌(PCa)是全球男性中发病率最高的恶性肿瘤。据预测,2026年美国PCa新增病例将达333 830例[1]。PCa发病风险与年龄高度相关,老年男性群体的患病及死亡风险显著升高。全球人口老龄化进程的加速,进一步加剧了全球医疗保健体系的负担。早期PCa的主要治疗手段包括手术切除及局部放化疗,这类方案通常可有效遏制肿瘤进展;随着Gleason分级的升高,雄激素剥夺疗法(ADT)逐渐成为主导治疗方案。然而,ADT治疗易诱导疾病进展为转移性去势抵抗性前列腺癌(mCRPC),这给PCa治疗带来了更大挑战。
雄激素受体(AR)作为类固醇激素的关键受体,是PCa发生发展的核心驱动因子。当前,以ADT及雄激素受体信号抑制剂(如恩杂鲁胺、阿帕鲁胺)为代表的PCa主流治疗策略,均通过靶向AR信号通路发挥抑癌作用,并已取得一定疗效。然而,多数患者在治疗后不可避免地产生耐药性,进而进展为更难治的去势抵抗性前列腺癌(CRPC)[2]。耐药性产生及AR持续激活仍是实现疾病长期有效控制的主要障碍,因此迫切需要开展更精准、深入的机制研究,并研发新型药物以克服耐药,改善患者治疗结局[3]。
铁死亡被认为是PCa治疗的潜在有效策略,尤其适用于晚期PCa[4]。铁死亡是一种铁离子依赖的、以脂质过氧化物累积为核心特征的程序性细胞死亡方式。与凋亡、坏死等经典细胞死亡方式不同,铁死亡的发生依赖于铁稳态、脂质代谢及抗氧化系统等细胞代谢途径,并受这些途径的精密调控。肿瘤细胞代谢异常活跃,会产生大量活性氧(ROS),且对铁离子需求增加,这使其对铁死亡的敏感性显著增强[5-6]。本文系统阐述AR对铁死亡的转录调控作用,总结并探讨近年来新发现的AR-铁死亡轴相关分子机制,评估天然产物靶向AR-铁死亡轴的临床应用潜力,并提出天然产物靶向AR-铁死亡轴有望成为克服PCa耐药的新型治疗范式。
1铁死亡
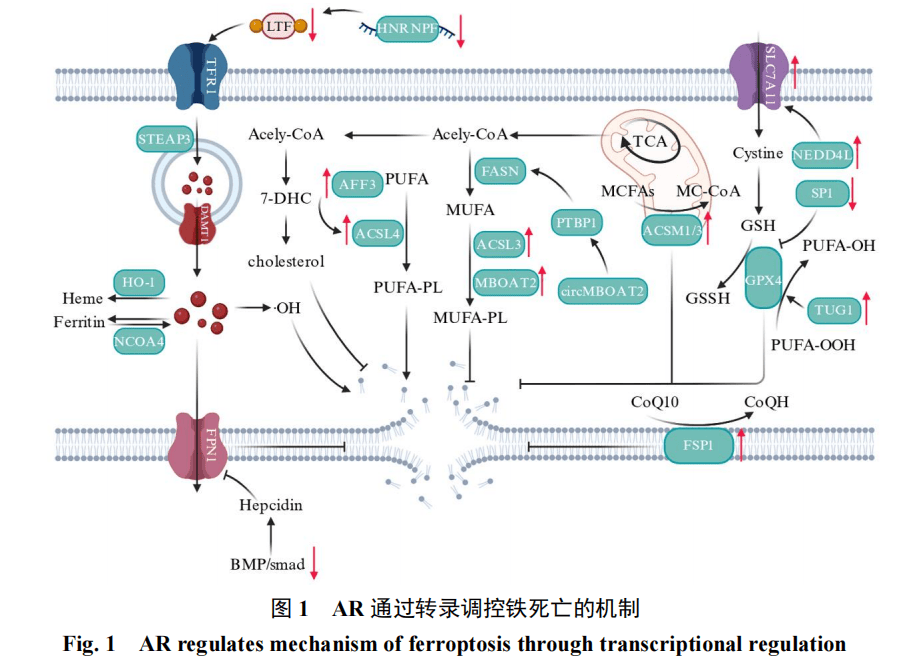
铁死亡是一种铁依赖的独特细胞死亡方式,其核心特征为脂质过氧化累积与抗氧化系统功能失调。铁离子是驱动铁死亡发生的关键因素:不稳定铁离子在细胞内蓄积后可触发芬顿反应,产生的大量自由基易攻击膜磷脂中的不饱和脂肪酸,导致细胞膜及线粒体膜脂质过氧化物异常堆积,最终诱导铁死亡发生。细胞内铁离子的转运过程具有明确的调控路径:主要由转铁蛋白及其受体转铁蛋白受体1(TFR1)介导进入细胞,内体中的Fe3+首先被金属还原酶(STEAP3)还原为Fe2+[7],随后经二价金属转运蛋白1(DMT1)转运至胞质并发生氧化反应;未被氧化的Fe²⁺则通过细胞膜上的膜铁转运蛋白(FPN1)回流至循环系统。铁调素作为胞外关键铁调节蛋白,可通过抑制FPN1的表达阻断铁离子向胞外转运,导致胞内铁离子蓄积,进而促进铁死亡发生[8]。
脂质过氧化是铁死亡的标志性事件,由多不饱和脂肪酸(PUFA)构成的膜磷脂(PUFA-PL)是脂质过氧化的核心底物。长链酯酰辅酶A合成酶4(ACSL4)是PUFA-PL合成的关键酶,可催化PUFA与辅酶A结合生成脂酰辅酶A;脂酰辅酶A进一步在溶血磷脂酰胆碱酰基转移酶3催化下与溶血磷脂酰胆碱结合,生成PUFA-PL并嵌入细胞膜磷脂层[9]。与之相对,参与磷脂合成的单不饱和脂肪酸(MUFA)具有抗氧化活性,其形成的MUFA-PL占比升高可显著增强细胞对铁死亡的抗性[10]。胆固醇作为细胞膜的主要组成成分,参与调控膜流动性及铁死亡进程:角鲨烯环氧化酶是胆固醇从头合成的关键调控酶,可通过促进胆固醇合成、升高胞内胆固醇含量抑制铁死亡发生[11];7-脱氢胆固醇作为胆固醇合成的中间代谢产物,可在7-脱氢胆固醇还原酶催化下转化为胆固醇,已有研究证实其是铁死亡的内源性抑制因子[12]。
抗氧化系统功能失调是铁死亡发生的重要助推因素。谷胱甘肽过氧化物酶4(GPX4)是经典的铁死亡抑制因子,可将氧化磷脂还原为无毒的脂醇,其活性严格依赖谷胱甘肽(GSH)。溶质载体家族7成员11(SLC7A11)作为关键的谷氨酸转运蛋白,可促进GSH从头合成,进而间接增强GPX4活性。铁死亡抑制蛋白1(FSP1)是定位于细胞膜的还原型辅酶Ⅱ依赖性氧化还原酶,其介导的抗氧化途径不依赖GPX4[13]:FSP1可催化细胞膜上的氧化型辅酶Q10(泛醌)转化为还原型辅酶Q10(泛醇),而泛醇作为具有抗铁死亡活性的抗氧化剂,可直接清除氧化应激产生的ROS。二氢乳清酸脱氢酶的功能与FSP1类似,其定位于线粒体内膜,可催化辅酶Q10转化为泛醇,直接中和线粒体产生的ROS以抑制铁死亡,且二氢乳清酸脱氢酶介导的抗氧化途径与GPX4、FSP1途径呈平行关系[14]。
2 AR-铁死亡轴介导的PCa耐药机制
AR作为促进PCa发生发展的关键转录因子,经配体激活后可转移至细胞核内参与转录调控。近年来,越来越多的研究证实,AR与铁死亡之间存在潜在关联。例如,在CRPC的体内、外模型中,采用铁死亡诱导剂erastin处理后,AR及其剪切变体AR-V7的表达水平显著下调[15]。核因子E2相关因子2(NRF2)是调控抗氧化防御系统的关键因子,可抑制PCa向CRPC的转化;而NRF2诱导剂能够通过促进AR及AR-V7的降解,增强恩杂鲁胺耐药性PCa对恩杂鲁胺的敏感性[16]。上述研究结果表明,AR与铁死亡之间存在相互调控关系。
2.1 AR与铁代谢的串扰在铁稳态调控及铁死亡抑制中的作用
铁稳态失衡引发的铁离子蓄积是驱动铁死亡的关键因素。相较于AR缺陷前列腺癌细胞(如DU145、PC3细胞系),AR阳性细胞对铁离子更敏感,AR与铁代谢通路间的相互调控可影响铁死亡进程[17]。转铁蛋白受体1可正向调控铁离子转运过程,促进铁离子进入细胞;而雄激素剥夺后,转铁蛋白受体1表达显著上调,进而导致细胞内Fe2+水平及ROS水平升高[18]。
近期研究发现,乳转铁蛋白(LTF)的表达受AR调控:AR可通过破坏乳转铁蛋白增强子(LTFe)与异质核核糖核蛋白F的相互作用,下调LTF的转录表达,从而抑制铁转运过程并阻碍铁死亡发生[19]。此外,铁调素高表达与前列腺特异性抗原水平呈正相关,可上调肿瘤增殖及存活相关标志蛋白的表达,而AR可通过调控铁调素表达抑制铁死亡。研究表明,AR可通过抑制骨形态发生蛋白-Sma和Mad相关蛋白信号通路,下调肝脏铁调素基因表达[20],进而稳定并上调FPN1水平[21]。
值得注意的是,铁代谢相关蛋白亦可反向调控AR表达。细胞内亚铁离子可通过血红素分解产生,血红素氧合酶-1(HO-1/HMOX1)是催化该过程的关键酶,其可通过调控信号转导和转录激活因子3抑制AR转录活性[22]。核受体共激活因子4作为铁自噬相关蛋白,同时也是AR的重要共激活因子,可与AR直接结合并促进其转录表达[23]。综上,AR与铁代谢通路的串扰可减少细胞内不稳定铁离子的蓄积,从而抑制铁死亡进程。
2.2AR调控PCa脂代谢通路及其对铁死亡的影响
AR作为代谢调控的关键转录因子,可通过增强PCa的脂质合成与脂肪酸氧化过程,推动癌细胞增殖。PUFA-PL是铁死亡的重要底物,其不饱和双键易成为ROS的攻击靶点。ACSL4介导的PUFA活化过程,能促进PUFA嵌入细胞膜,进而驱动铁死亡发生。
ALF转录延伸因子3(AFF3)是AR调控的核转录激活因子,AR可抑制AFF3的转录活性。研究表明,AFF3能够通过调控ACSL4的活性,升高ROS及丙二醛(MDA)水平,最终诱导铁死亡[24]。此外,AR与ACSL4存在直接互作关系,可通过结合ACSL4的启动子区域抑制其转录表达,减少脂质过氧化物的积累[25]。
ACSL家族的另一成员长链酯酰辅酶A合成酶3(ACSL3)同样受AR转录调控,但ACSL3在催化PUFA活化方面的效能弱于ACSL4。ACSL3对饱和脂肪酸(SFA)和单不饱和脂肪酸(MUFA)具有更高亲和力,可促进MUFA-PL生成,从而抑制铁死亡[25]。MUFA及MUFA-PL的积累能够减少脂质过氧化反应及ROS产生,具备抗氧化活性。
膜结合O-酰基转移酶域包含2(MBOAT2)属于酰基转移酶,具有LPCAT3酶活性,可促进MUFA转化为溶血磷脂酰乙醇胺。近年研究发现,雄激素可调控MBOAT2的表达,AR通过增强MBOAT2的转录活性,促进MUFA嵌入膜磷脂,进而抑制铁死亡发生[26]。值得注意的是,MBOAT2介导的抗铁死亡效应不依赖于GPX4。
CircMBOAT2是一类与脂质代谢相关的环状RNA(circRNA),参与调控脂质从头合成过程。聚嘧啶束结合蛋白1(PTBP1)为核糖体蛋白,CircMBOAT2可通过与PTBP1直接结合稳定其表达,进而间接调控脂肪酸合酶(FASN)的表达,促进脂肪酸合成及MUFA生成[27]。
脂肪酸氧化过程在PCa中异常活跃,近期研究发现中链脂肪酸氧化可抑制铁死亡发生。酰基辅酶A合成酶中链家族成员1(ACSM1)和成员3(ACSM3)是负责中链脂肪酸活化的脂酰酶,AR通过上调ACSM1/3的表达,促进中链脂肪酸的β-氧化,从而抑制铁死亡[28]。
胆固醇是雄激素合成的重要前体,胆固醇合成可促进AR激活并增强其转录活性,进一步负向调控铁死亡,推动PCa进展及耐药发生。固醇调节元件结合蛋白(SREBP)是胆固醇合成的关键调节因子,抑制SREBP表达可阻断雄激素生成及AR信号传导[29]。综上,AR与脂代谢之间的相互作用可通过减少脂质过氧化底物积累,阻碍铁死亡发生。
2.3 抗氧化系统调控铁死亡的作用机制
抗氧化系统的功能缺失是驱动细胞铁死亡发生的关键因素。SLC7A11-GPX4通路是目前公认的经典抗氧化轴,AR可通过上调该通路的表达水平,进而发挥抑制铁死亡的作用。Sp1转录因子(SP1)隶属于Sp/KLF家族,参与调控细胞死亡进程。研究发现,AR可与SP1相结合形成复合物,促使GPX4从SP1上解离,进而提升GPX4的转录表达水平,最终减少细胞内脂质过氧化物的蓄积[30]。
值得注意的是,在非PCa相关疾病中,AR则可能通过抑制GPX4的活性,进而诱导铁死亡的发生。在一项针对良性前列腺增生的研究中发现,AR功能缺陷可促使巨噬细胞释放白细胞细胞介素(IL)-1β,进而促进长链非编码RNA(llncRNA)牛磺酸上调基因1(TUG1)的转录表达。TUG1表达水平的升高可通过与微小RNA-188-3p(miR-188-3p)竞争性结合,减弱miR-188-3p对GPX4的靶向抑制作用,最终实现对铁死亡的抑制[31]。
SLC7A11是细胞内谷氨酸的重要转运蛋白,其表达水平与GSH的合成密切相关,并可进一步增强GPX4的活性。NEDD4样E3泛素蛋白连接酶是一类重要的E3泛素连接酶,AR可通过诱导NEDD4样E3泛素蛋白连接酶的表达,进而上调SLC7A11的水平,增强谷氨酸转运能力与GPX4酶活性,发挥抑制铁死亡的效应[32]。
在CRPC中,全长雄激素受体(AR-FL)与AR-V7均可直接结合于SLC7A11的启动子区域,发挥抗铁死亡作用。尤为值得关注的是,AR-V7与SLC7A11启动子的亲和力显著高于AR-FL[33]。
FSP1是一类能够催化泛醌生成泛醇的氧化还原酶,其介导的抗氧化通路与GPX4通路相互独立。AR亦可通过调控FSP1的表达,减少细胞内ROS的蓄积,从而阻断铁死亡的发生进程[34]。上述研究结果表明,AR主要通过增强细胞抗氧化活性,实现对铁死亡的抑制作用。
AR通过转录调控铁死亡作用机制总结见图1。
3 靶向AR-铁死亡的天然产物
靶向AR-铁死亡信号轴治疗PCa具有较高的应用潜力,然而化学药的耐药性问题,仍是制约其临床疗效的主要瓶颈。近年来,诸多研究相继发掘出可用于治疗难治性前列腺癌的天然产物,并阐明了其潜在分子机制。根据作用靶点的差异,这类天然产物可划分为3大类别,即直接靶向AR-铁死亡轴、靶向PCa AR信号通路以及靶向PCa细胞铁死亡过程。
3.1 直接靶向AR-铁死亡轴的天然产物
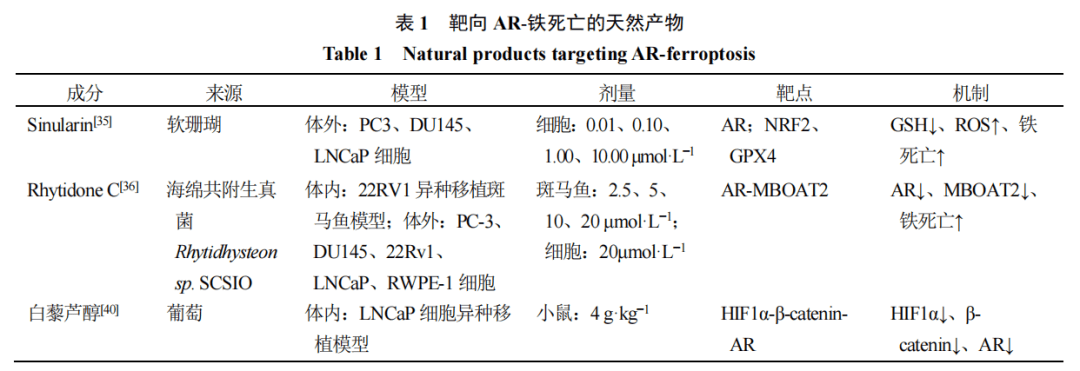
Sinularin是一种分离自海洋珊瑚的天然活性产物。研究证实,该化合物可通过抑制AR表达及其下游信号传导,发挥抑制前列腺癌细胞增殖与迁移的作用。除此之外,Sinularin还能显著下调Nrf2和GPX4的表达水平,削弱细胞的抗氧化能力,进而促进铁死亡发生[35]。不过,该研究并未进一步探究Sinularin是否能够调控AR与Nrf2/GPX4之间的信号串扰。已有研究表明,AR可通过直接抑制GPX4表达诱导铁死亡,这也提示Sinularin或许是通过调控AR信号通路,进而抑制GPX4表达,最终实现对铁死亡的调控。
Rhytidone C是海绵共附生真菌Rhytidhysteonsp. SCSIO 41423的次级代谢产物。研究发现[36],该化合物可通过调控AR-MBOAT2信号轴抑制前列腺癌进展,且22Rv1细胞异种移植斑马鱼模型的实验结果也验证了其抗前列腺癌活性。具体而言,分子对接、免疫荧光等实验结果显示,Rhytidone C与AR具有较强的结合亲和力,能够显著降低AR的蛋白表达水平,并抑制其核转位能力。进一步研究表明,Rhytidone C处理22Rv1前列腺癌细胞后,AR下游靶基因的mRNA及蛋白表达水平均显著下调,其中包括铁死亡抑制因子MBOAT2;同时,透射电镜观察到细胞线粒体出现皱缩、线粒体嵴消失等铁死亡的典型形态学特征。
3.2靶向AR信号通路的天然产物
阿曲酸是一种分离自非洲Pygeum africanum树皮的天然产物。研究表明[37],阿曲酸可通过3方面抑制AR活性:①与AR竞争性结合,阻断AR的配体依赖性激活;②抑制AR核易位;③抑制AR与雄激素反应元件(ARE)的结合,降低AR转录活性及促血管生成因子血管生成素2(ANGPT2)的转录表达。除阿曲酸外,其他多种天然产物也可通过靶向AR激活的不同环节下调AR表达,进而抑制PCa进展。例如,源自万寿菊的α-Terthienyl可通过抑制双氢睾酮(DHT)与AR结合并阻断其核易位,抑制AR阳性PCa细胞(LNCaP、22Rv1)的增殖;在LNCaP细胞异种移植小鼠模型中,α-Terthienyl干预可显著降低肿瘤体积及质量[38]。Jiang等[39]的研究证实,源自传统中药的黄芩素可通过调控AR启动子−698~−412 bp区域的活性,降低AR转录水平,从而抑制PCa进展。Mitani等[40]则发现,白藜芦醇可通过下调HIF-1α的表达,阻断AR辅助活化因子β-catenin的核积累及AR转录激活过程,最终实现AR表达下调与PCa抑制的双重效应。
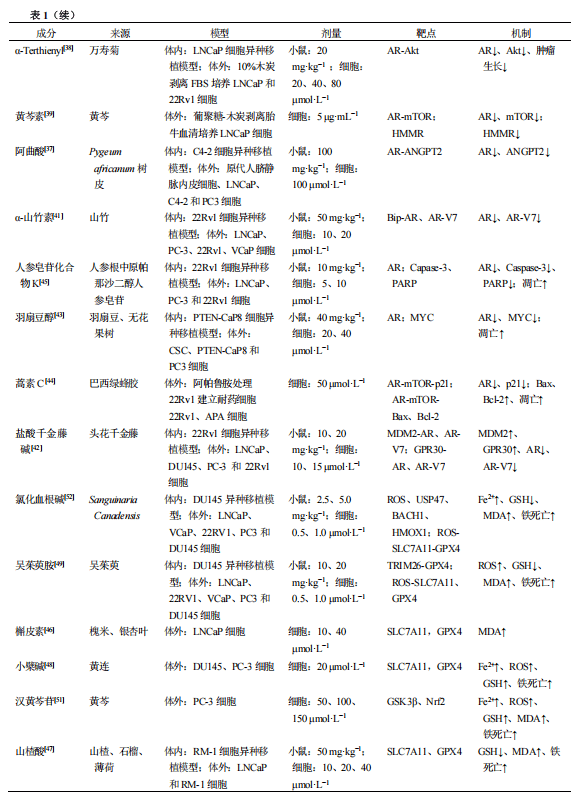
α-山竹素是从山竹中分离得到的黄酮类化合物。研究证实[41],该化合物可促进AR-FL及AR-V7的降解,对CRPC进展具有显著抑制作用。具体机制为:α-山竹素处理可上调内质网伴侣蛋白免疫球蛋白重链结合蛋白(Bip)的表达,同时促进Bip与AR-FL、AR-V7的相互结合,进而诱导二者发生泛素化修饰并加速降解。
盐酸千金藤碱为千金藤碱的成盐衍生物,是源自传统中药头花千金藤中的生物碱类成分。研究证实[42],盐酸千金藤碱对AR阳性PCa细胞(LNCaP、22Rv1)具有较强毒性,其机制为通过增强E3泛素连接酶MDM2的活性,促进AR及AR-V7降解。此外,该研究还发现,G蛋白偶联雌激素受体30(GPR30)作为盐酸千金藤碱的另一作用靶点,同样可介导AR降解。研究者通过构建22Rv1细胞异种移植小鼠模型,进一步验证了盐酸千金藤碱对AR及AR-V7的抑制效应。该研究明确了天然产物可通过多靶点调控AR降解以抑制PCa进展的药理特性。
羽扇豆醇属于三萜类化合物,具备AR抑制活性,可增强癌细胞对恩杂鲁胺的敏感性,具体表现为促进癌细胞凋亡、抑制细胞增殖[43]。此外,天然产物在体内的代谢转化特性,为其实现AR靶向作用提供了更多可能性。蒿素C是源自于巴西绿蜂胶的酚类化合物。在构建的AR抑制剂阿帕鲁胺耐药细胞系22Rv1/APA中,蒿素C通过阻碍AR与其配体DHT结合,极大抑制AR激活[44]。这一作用增强了PCa对阿达帕胺的敏感性,凸显其在克服PCa耐药中的优势。
人参皂苷化合物K是人参皂苷在体内的代谢转化产物,属于源自传统中药人参的四环三萜类化合物。Jiang等[45]研究发现,人参皂苷化合物K处理可使LNCaP及22Rv1细胞中AR与AR-V7的表达水平显著下调,从而抑制癌细胞生长。具体机制为:人参皂苷化合物K可降低AR启动子活性,而非通过增强AR蛋白修饰实现调控;同时,AR-V7及其靶基因UBE2C、EDN2的转录水平也显著降低。该研究证实,天然产物的体内代谢产物在PCa耐药治疗中具有较高的应用潜力。
3.3靶向铁死亡的天然产物
槲皮素属于黄酮醇类化合物,广泛存在于槐米、银杏叶等植物中。王君君等[46]研究发现,采用槲皮素处理LNCaP细胞后,细胞增殖受到显著抑制,同时MDA水平升高,SLC7A11与GPX4的表达下调,提示槲皮素可能通过诱导铁死亡降低细胞活力,进而抑制PCa进展。在Hu等[47]的研究中,同样证明了源自于山楂的山楂酸的铁死亡诱导作用。山楂酸处理RM-1细胞后,受损线粒体增加、GPX4蛋白表达及SLC7A11的基因表达水平降低,表明山楂酸通过诱导铁死亡抑制PCa进展。
小檗碱是来源于黄连根茎的生物碱,既往研究证实其可通过诱导铁死亡抑制多种疾病进展,包括动脉粥样硬化、糖尿病肾病及PCa等。穆克飞等[48]研究表明,小檗碱可通过升高ROS水平及Fe²⁺含量诱导PCa细胞铁死亡。具体机制为:小檗碱处理PCa细胞DU145、PC3后,SLC7A11的基因及蛋白表达水平显著下调;而过表达SLC7A11可部分逆转小檗碱诱导的ROS升高及GSH耗竭。该研究在体外明确了小檗碱通过诱导铁死亡发挥抗肿瘤作用的分子机制。
吴茱萸胺是源自于吴茱萸的生物碱类化合物。Li等[49]研究发现,吴茱萸胺通过降低GPX4表达诱导PCa细胞铁死亡。具体机制为:吴茱萸胺处理PCa细胞后,ROS水平升高,导致SLC7A11与GPX4的蛋白表达水平下降;同时,吴茱萸胺降低E3泛素连接酶TRIM26表达,导致GPX4稳定性下降,进而增强脂质过氧化水平和诱导铁死亡。DU145细胞异种移植小鼠模型中也同样发现TRIM26、GPX4、SLC7A11的蛋白水平降低。该研究凸显天然产物多靶点诱导铁死亡抑制癌症进展的药物特性。
汉黄芩苷是来源于中药黄芩的黄酮类化合物。既往研究显示,黄酮类天然产物可通过抑制抗氧化酶活性、削弱细胞抗氧化能力,从而诱导铁死亡并抑制疾病进展,例如毛蕊异黄酮可通过抑制Nrf2通路诱导人甲状腺癌细胞铁死亡[50]。张丽君等[51]研究发现,汉黄芩苷也可通过调控Nrf2抑制PCa进展,具体机制为:在PCa细胞PC3中,汉黄芩苷通过抑制糖原合成酶激酶-3β的磷酸化,下调磷酸化GSK-3β及Nrf2的蛋白表达水平,最终诱导铁死亡。
氯化血根碱是苯并苯蒽啶生物碱类天然产物,可通过调控铁稳态诱导铁死亡。氯化血根碱处理后,HO-1介导的血红素分解过程增强,导致游离亚铁离子蓄积。研究证实[52],氯化血根碱可通过促进ROS生成抑制泛素特异性肽酶47(USP47)表达,导致与HO-1启动子结合的BTB域和CNC同系物1(BACH1)蛋白稳定性降低,进而增强HO-1的转录活性,最终促进血红素分解及铁蓄积,诱导铁死亡发生。
川楝素是来源于Melia toosendan树皮的四环三萜类化合物。Shen等[53]研究发现,川楝素处理DU145、LNCaP细胞后,Fe2+、MDA、脂质ROS水平显著增加,提示川楝素可以诱导PCa细胞铁死亡。具体机制为:川楝素降低泛素特异性蛋白酶39(USP39)表达,抑制USP39的去泛素化活性并降低polo样激酶1(PLK1)稳定性,促进铁死亡的发生。
靶向AR-铁死亡的天然产物及机制总结见表1。

4 天然产物抵抗PCa耐药临床转化挑战与对策
尽管近年来AR调控铁死亡的分子机制研究持续深入,但天然产物的临床转化仍面临严峻挑战。天然产物凭借多靶点调控、低毒性的独特优势,在靶向AR-铁死亡轴逆转PCa耐药方面展现出良好的治疗潜力,但其从基础研究向临床应用的转化却遭遇核心瓶颈。当前相关研究仍局限于“现象描述多于机制解析”“基础活性验证先于临床应用探索”的阶段,机制阐释的模糊性与临床转化的阻滞性,已成为制约天然产物攻克PCa耐药难题的核心症结。
首先是机制阐释的模糊性问题。现有研究大多证实天然产物可同时作用于AR信号通路与铁死亡通路,却未能厘清二者之间的因果调控关系。以Sinularin为例,该化合物可同时抑制AR表达并下调NRF2、GPX4的表达水平,相关研究仅观察到“AR表达受抑—NRF2/GPX4表达下调-铁死亡效应增强”的线性关联现象,却未能解答核心科学问题:软珊瑚素是否通过调控AR表达,进而下调GPX4或NRF2的表达水平以发挥促铁死亡作用[35]。此类关键实验数据的缺失,使得AR与铁死亡通路之间的调控机制长期处于模糊不清的状态。
为突破这一瓶颈,未来研究需引入基因编辑与功能干预实验,借助CRISPR-Cas9技术敲除AR基因,或过表达AR野生型及突变体,明确AR与铁死亡通路的上下游调控关系,界定AR调控铁死亡相关蛋白的具体功能结构域。这一策略不仅能够阐明AR-铁死亡通路的调控机制,更能为天然产物的精准靶向研发提供潜在候选靶点。
5结语与展望
AR与铁死亡的交互调控在PCa进展中发挥关键作用,靶向AR-铁死亡轴有望成为攻克PCa耐药的新型临床治疗策略。本文明确,AR可通过多维度转录调控网络调控铁死亡进程:在铁稳态调控中,AR通过抑制Fe²⁺内流(LTFe-LTF通路)与促进Fe²⁺外排(BMP/Smad通路),减少胞内不稳定铁池积累;在脂代谢层面,AR通过调控ACSL4、MBOAT2的转录活性,平衡脂质过氧化底物(多不饱和脂肪酸磷脂,PUFA-PL)与抗氧化脂质(单不饱和脂肪酸磷脂,MUFA-PL)的比例;在抗氧化防御系统中,AR不仅可直接结合SLC7A11启动子以增强SLC7A11-GPX4轴的抗氧化活性,还能调控FSP1介导的非GPX4依赖性抗氧化通路。最终,AR构建起“AR-铁死亡-耐药”的调控环路,为靶向干预提供了明确的机制基础。
本研究总结的3类天然产物展现出靶向AR-铁死亡轴的独特优势:第一类为直接靶向该轴的化合物,可实现AR信号通路与铁死亡的协同调控;第二类为AR靶向型化合物,为克服AR变异介导的耐药提供了潜在候选分子;第三类为铁死亡靶向型化合物,可从铁死亡执行环节突破耐药屏障。这些天然产物具备的“多靶点、低毒性”特性,为PCa耐药治疗提供了区别于化学药物的全新治疗范式。然而,当前天然产物靶向AR-铁死亡轴的研究仍面临亟待突破的瓶颈,主要包括机制探究深度不足、临床转化难度较大。未来需联合基因编辑、时空代谢组学等前沿技术,明确AR与铁死亡关键分子(GPX4、ACSL4)的上下游调控关系及核心作用结构域,实现核心成分的精准量化与给药剂量的优化。
综上,AR-铁死亡轴的发现为PCa耐药机制研究提供了全新视角,而天然产物对该轴的靶向潜力则为抗PCa药物研发开辟了新方向。后续通过机制研究的精细化、成分研发的精准化及转化研究的系统化推进,有望为晚期PCa患者提供安全有效的新型治疗方案,为破解PCa耐药难题提供关键支撑。